Work flow¶
Building the database¶
The current version of scConnect have version 2019-5 of the latest ligand, receptor and interaction data from guide to pharmacology allready compiled.
Should you find the need to change to an older database version, download and replace the csv files under data/GTP_tables/
and run scConnect.database.setup_database(). If you have allready built this database, set the version using scConnect.database.version
Working with AnnData objects and Scanpy¶
scConnect rely heavily on the workflow and data structures included in the scRNA-seq package scanpy. Please refer to their documentation for working with single cell data and how to detect cell populations.
To run the functions in this package, you will need an AnnData object with any kind of cell population groups under adata.obs.
Make gene calls¶
In order to link gene expression to precence or absence of ligands and receptors for cell populations, we must decide how to measure gene expression on a cell population basis. There are several algorithms to do this:
Using threshold parameters
scConnect.genecall.betabinomialTrinarization |
|
scConnect.genecall.meanThreshold |
|
scConnect.genecall.medianThreshold |
Non-parametric
scConnect.genecall.percentExpression |
|
scConnect.genecall.meanExpression |
Calculating ligand scores for peptide ligands are staight forward, as this can be equal to the gene call. Calculating molecular ligands involves more genes, as synthesis, vesicle transporters and reuptake transporters are all nesesary to produce and transport the ligand. Further more, some genes cannot be expressed as these whould convert the ligand to another ligand, these we call exclusion genes.
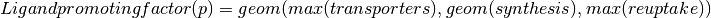
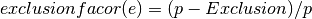
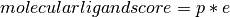
Optimizing threshold settings for neurons¶
One common feature of neuronal tissue is that it contain cells that are either glutamatergic or gabaergic, but they do rarle express both neurotransmitters. The neurons hence segregate into excitatory or inhibitory phenotypes. This feature can be used to find the lowest threshold that replicates this biological feature, and can be used as a guide when selecting a threshold.
The effect on this segregation can be assesed for all thresholds using scConnect.genecall.optimize_segregation() (only testing betabinomial trinarization).
Select the lowest threshold value producing the highest segregation score and run scConnect.genecall.betabinomialTrinarization() with this threshold.
Visualize the segregation using scConnect.genecall.plot_segregation().
Deciding what to connect¶
After running the gene call algorithm of your choice, it is time to link the gene calls to ligands and receptors. To do this, run the following functions on all datasets that you want to investigate connectivity between.
scConnect.connect.ligands |
|
scConnect.connect.receptors |
Interactions between cell populations are found using scConnect.connect.interactions().
It expects two datasets, one emitter and one target dataset.
If you are interested in the interactions between cell populations in just one dataset,
supply this dataset as both emitter and target.
The end goal is to build a graph representing all the interactions that occur between our cell populations. To build a graph, we need a list of all interactions, here named an edge list, and also information about the cell types, here named node list.
scConnect.connect.interactions() returns a python list containing edge information,
and several edge lists can be added together using the + operator.
Warning
Cell population names has to be unique between different datasets and will otherwise be considered the same population in the graph.
It is usefull to export information about ligand and receptor expression for cell populations
from all datasets before building a connectivity graph. This is done by supplying a list of all datasets to
scConnect.connect.nodes(). This will provide a list of all nodes with relevant metadata.
Building the graph¶
The graph is constructed using scConnect.graph.build_graph() with the edge list and node list as arguments.
When the graph is constructed some edge metrics are automatically calculated:
scConnect.graph.loyalty |
|
scConnect.graph.promiscuity |
|
scConnect.graph.weighted_score |
The graph is a networkX multi directional graph, with each ligand receptor pair constituting an edge between two nodes. For further information on how to work with the graph, please refer to networkX documentation
Analysing the graph¶
to be continued..